
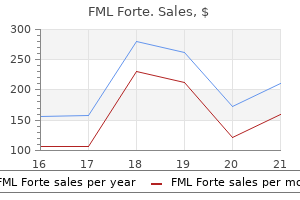
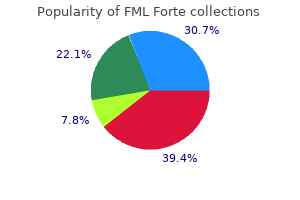
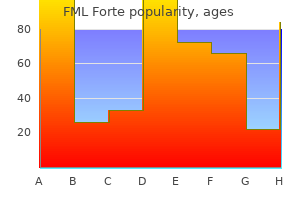
FML Forte
FML Forte dosages: 5 ml
FML Forte packs: 1 suspensions, 2 suspensions, 3 suspensions, 4 suspensions, 5 suspensions, 6 suspensions, 7 suspensions, 8 suspensions, 9 suspensions, 10 suspensions

Fml forte 5 ml cheap with mastercard
Treatment and consequence Holocarboxylase synthetase deficiency may be handled successfully with pharmacological doses of biotin allergy treatment 4 addiction cheap 5 ml fml forte otc. The required dose of biotin is dependent on the severity of the enzyme defect and has to be 12 allergy forecast west palm beach generic fml forte 5 ml visa. In spite of apparently full recovery, biochemical and clinical abnormalities persist in some patients owing to the high Km for biotin within the faulty holocarboxylase synthetase. The prognosis is sweet if therapy is initiated immediately, except for affected individuals with Km variants. Clinical presentation Patients with d-2-hydroxyglutaric aciduria exhibit variable phenotypes. They have been divided into two subgroups primarily based on scientific, neuroradiological, and molecular findings. Patients with d-2-hydroxyglutaric aciduria kind I are moderately affected and often comply with a light medical course with variable symptoms including learning difficulties, muscular hypotonia, and macrocephaly. Patients present with encephalopathy of early childish onset, demonstrating a mix of catastrophic epilepsy, muscular hypotonia, cerebral visible failure, and severe psychomotor retardation. Neuroimaging findings in these patients show ventriculomegaly, enlarged subarachnoid spaces, subdural effusions, subependymal cysts, and delayed cerebral maturation. Recently, agenesis of the corpus callosum, bilateral involvement of the striatum, and cerebral artery infarctions had been added to the spectrum. Diagnosis the biochemical hallmark of this illness is the buildup of d-2-hydroxyglutaric acid in all body fluids. Demonstration of elevated ranges of 2-hydroxyglutaric acid must be adopted up by differential quantitation of the two isomers l- and d2-hydroxyglutaric acid. The lateral ventricles are highly dilated, occipital more than frontal, the cerebral maturation is delayed. However, due to attribute further parameters these differential diagnoses are usually simple to exclude. Long-term care of patients should entail common analysis of cardiomyopathy and the progression of neurological disease. Severely affected youngsters might die in infancy, while moderately affected patients have a better prognosis as a lot as an unimpaired life. The disease is attributable to deficiency of the flavin adenine dinucleotide-dependent mitochondrial enzyme l-2hydroxyglutarate dehydrogenase changing l-2-hydroxyglutarate to 2-oxoglutarate. Clinical presentation l-2-Hydroxyglutaric aciduria was first described by Duran and coworkers in 1980. In the first 2years of life, mental and psychomotor growth could also be normal or slightly delayed. Febrile seizures, nonspecific developmental delay, and muscular hypotonia are the presenting signs. Progressive ataxia, variable extrapyramidal and pyramidal signs, epilepsy, and progressive studying difficulties ultimately develop. The neuroimaging findings in l-2-hydroxyglutaric aciduria are distinctive and principally uniform comprising a progressive lack of arcuate fibres mixed with progressive cerebellar atrophy and sign adjustments in globus pallidus and the dentate nuclei. Diagnosis l-2-hydroxyglutaric aciduria leads to a somewhat homogeneous clinical picture and characteristic abnormalities on neuroimaging. Prenatal diagnosis relies on the evaluation of l-2-hydroxyglutaric acid in amniotic fluid samples or molecular evaluation. The oldest recognized sufferers are close to 40years of age, bedridden, and severely disabled. Combined d-2- and l-2-hydroxyglutaric aciduria Aetiology/pathophysiology the molecular basis of mixed d-2- and l-2-hydroxyglutaric aciduria has recently been unravelled. This includes epileptic encephalopathy, muscular hypotonia, respiratory insufficiency, extrapyramidal motion disorders, cortical visual failure, microcephaly, and severe developmental delay. Subcortical white matter is severely deficient with a lot much less involvement of the internal capsule and the periventricular white matter. Patients with a severe onset and intractable epileptic seizures have a poor prognosis: eight of twelve recently reported instances died between 1 month and 5 years of age. In 1986, it was acknowledged that N-acetylaspartic aciduria was attributable to deficient aspartoacylase in a toddler with a similar medical presentation. E285A within the aspartoacylase gene, localized on 17p13-pter, accounts for greater than 80% of alleles in Ashkenazi Jews and for 60% of alleles in sufferers of non-Jewish origin. In healthy individuals, excessive concentrations of N-acetylaspartic acid (8mmol/g tissue) are completely found in brain tissue. Aspartoacylase is localized in oligodendrocytes catalysing the deacetylation of N-acetylaspartic acid to produce acetate, a substrate for the synthesis of myelin lipids together with cholesterol. It has been proposed that N-acetyl-l-aspartate might operate as a molecular water pump in myelinated neurons, transporting water against its gradient from neurons to oligodendrocytes. Hypotonia with distinguished head lag, epilepsy, lack of beforehand acquired expertise, as properly as progressive megalencephaly are frequently found. As the disease progresses, affected kids develop pyramidal indicators, and eventually decerebration. Neuroimaging reveals characteristic symmetrical leukodystrophic modifications with lack of arcuate fibres; histology demonstrates spongiform degeneration, specifically of the cortex and subcortical white matter. In infancy, modifications could also be delicate and misinterpreted as delayed myelination or periventricular leukomalacia. Note the marked discrepancy between the severely affected subcortical white matter and the relatively spared central white matter, at least frontally. Ethylmalonic encephalopathy Aetiology/pathophysiology Ethylmalonic encephalopathy is a devastating, infantile, autosomal recessive neurometabolic dysfunction affecting the brain, gastrointestinal tract, and peripheral veins. Only lately, it was elucidated using Ethe1-deficient mice that the poor protein is a mitochondrial sulphur dioxygenase which is involved within the catabolism of sulphide in ethylmalonic encephalopathy. As a consequence, toxic levels of sulphide and thiosulphide are found causing powerful inhibition of cytochrome c oxidase, short-chain fatty acid oxidation, and exerting vasoactive and vasotoxic effects. This explains poor mitochondrial energy metabolism, the irregular accumulation short-chain natural acids, acylglycines and acylcarnitines, as nicely as microangiopathy. Clinical presentation Ethylmalonic encephalopathy is characterised biochemically by ethylmalonic aciduria and methylsuccinic aciduria, lactic acidaemia, and clinically by extreme psychomotor retardation, acrocyanosis, petechiae, and chronic diarrhoea. Diagnosis the biochemical hallmark is increased urinary excretion of ethylmalonic and methylsuccinic acids associated with abnormal excretion of C4- and C5- (n-butyryl-, isobutyryl-, isovaleryl-, and 2methylbutyryl-) acylglycines and acylcarnitines as well as intermittent lactic acidosis. Since major mitochondrial issues are an necessary differential analysis, enzymatic analyses of respiratory chain enzymes in muscle biopsy specimen have been carried out in some sufferers revealing secondary cytochrome c oxidase deficiency. Increased ethylmalonate in urine is also present in multiple- and short-chain acyl-CoA dehydrogenase deficiencies, main respiratory chain deficiencies, and Jamaican vomiting illness. The prognosis is poor and ethylmalonic encephalopathy is usually lethal in early childhood. The asterisked enzymes are 1, phenylalanine hydroxylase; 2, tyrosine hydroxylase; 3, dihydrobiopterin reductase; four, tyrosine aminotransferase; 5, homogentisic acid oxidase; 6, fumaryl acetoacetate hydrolyase; and 7, tryptophan hydroxylase.
Buy discount fml forte 5 ml on-line
This is the results of a lack of inhibition with resultant elevated contraction of involved muscle tissue allergy testing vaughan purchase fml forte 5 ml. In people allergy keywords cheap fml forte 5 ml with visa, the flexor muscle tissue of the upper extremity and extensor muscles of the decrease extremity are primarily involved as demonstrated by hemiparetic individuals with the lower extremity prolonged (which could be an help to ambulation) and higher extremity flexed on the elbow and wrist, and sometimes held throughout the abdomen. Persistent untreated spasticity can finally result in contractures with resultant immobility of the concerned joints. Spasticity needs to be distinguished from the rigidity seen in affiliation with some extrapyramidal issues. Thus, the acquainted clinical presentation of 584 Encyclopedia of the Neurological Sciences, Volume four doi:10. This pattern is unlike that of spasticity, which, as said above, primarily impacts flexor muscles of the upper extremity and extensor muscular tissues of the decrease extremity. It is mostly most simply demonstrated on the ankle, though it can be detected on the patella and sometimes in the higher extremity. It outcomes from alternating and involuntary muscular contracture and leisure in speedy succession. Clonus can be spontaneous and generated by routine movements of an involved extremity. It generally occurs when a person retires to bed with related voluntary stretching of the paretic spastic limb. Although the Babinski signal is one of the best recognized, there are a number of different methods to generate an upgoing toe which would possibly be manifestations of the activation of heightened withdrawal reflexes much like the heightened deep tendon reflex. Associated problems usually include aphasia, dysphagia, contralateral hemisensory disturbances, and contralateral visual field loss. In this setting, the absence of associated signs and signs helps to define the placement of the lesion. Given the small diameter of the spinal wire, extraaxial, as well as intraaxial, lesions causing compression often impact the corticospinal tracts bilaterally. This can result in quadriparesis with cervical wire lesions or paraparesis with thoracic cord lesions. The distribution of the sensory loss can be extremely helpful in localizing the extent of a lesion. In this setting, a person will have ipsilateral weak spot and proprioception loss, along with contralateral lack of pain and temperature sense. Also, important is recognizing the occasional individual with psychogenic weak spot in the type of a conversion reaction or malingering. In the case of hemiparesis, a cautious history and physical examination can typically result in the proper diagnosis. The affected person with psychogenic hemiparesis will have normal reflexes, no Babinski signal or clonus, and normal tone. The sufferers are then asked to press their heel downward, as strongly as attainable. Definition Uremia is a condition brought on by renal failure and extreme accumulation of products of protein metabolism, as nicely as loss of intrinsic kidney homeostatic and endocrine perform. Caution must be used with renally excreted antiepileptic medication, for example, levetiracetam and gabapentin. Pathogenesis of Uremic Encephalopathy Clinical Manifestations of Acute and Chronic Uremic Encephalopathy Uremic encephalopathy could be divided into acute and persistent varieties, although there are considerable overlaps. Some of the most probably chemical substances include guanidino compounds, parathyroid hormone, urea, and various center molecules. Encephalopathy Related to Treatment of Uremia Acute Uremic Encephalopathy Acute uremic encephalopathy is a florid sickness starting from subtle executive dysfunction to coma. Delirium, both hypoactive or hyperactive, is the primary stage with fluctuating, decreased consideration, impaired construction and writing, executive dysfunction, behavioral changes, and sleep disturbances. Hyperventilation, with Kussmaul respiration, occurs during times of metabolic acidosis. Motor findings embrace paratonia, multifocal myoclonus, action myoclonus, stimulussensitive myoclonus, tremor, asterixis, and seizures. Uremic coma, now unusual, is typically accompanied by Kussmaul respiration associated to metabolic acidosis. Dialysis Disequilibrium Dialysis disequilibrium, affecting patients during or shortly after hemodialysis, is caused by shifts of water, causing cerebral edema. Headache, nausea, vomiting, restlessness, myoclonus, disorientation, and somnolence are the main options. More severe manifestations embrace natural psychosis, generalized seizures, stupor, or coma. In addition, the anorexia, nausea, and vomiting, present in some patients with chronic uremia, might lead to dramatic discount in oral thiamine consumption. As in acute uremic encephalopathy, postural or kinetic tremor, myoclonus, and asterixis can happen. With improved remedy methods, Encyclopedia of the Neurological Sciences, Volume 4 doi:10. Furthermore, microhemorrhages and infarcts are often seen in addition to infarction. Finally, cortical gray matter, along with underlying white matter, can be involved. In common, remedy is focused at managing hypertension, withdrawal of the offending agent, and treating the underlying condition. Although much less common with aggressive dialysis, frontally predominant triphasic waves with a frontooccipital gradient could be seen in severe azotemia or in decompensated chronic renal failure. They do, nevertheless, generally subside and will disappear after successful renal transplantation. Of course, a variety of the therapy modalities have their very own issues and require remedy. Introduction the vagus nerve (cranial nerve X) is a mixed nerve that originates within the medulla and accounts for a lot of the visceral innervation of the physique. The rootlets unite to type the vagal nerve trunk, which leaves the skull via the jugular foramen, throughout the identical dural sleeve because the glossopharyngeal and spinal accessory nerves. The vagus provides multiple branches at the stage of the neck, thorax, and stomach. Motor fibers leave the cervical vagus as three branches: pharyngeal, superior laryngeal, and inferior laryngeal (recurrent nerve). Both vagal nerves communicate with branches of the cervical sympathetic trunk, forming a mixed community of preganglionic parasympathetic and postganglionic sympathetic fibers. The pharyngeal department innervates all muscles of the pharynx, except the stylopharyngeus (supplied by the glossopharyngeal nerve) and tensor veli palatini (supplied by the trigeminal nerve). The superior laryngeal nerve subdivides an external branch that innervates the cricothyroid muscle and an inside department that gives sensory innervation to the pharynx. The recurrent laryngeal nerves supply all laryngeal muscular tissues besides the cricothyroid muscle.
Syndromes
- Frequent or severe infections
- Toxic shock syndrome
- Celiac disease
- Support groups (such as Alcoholics Anonymous)
- Pale skin color (pallor)
- Shortness of breath or lightheadedness
- Your injury does not appear to be getting better with time
Fml forte 5 ml order visa
In alkaptonuria nitisinone best allergy medicine for 7 year old fml forte 5 ml order on line, as predicted bread allergy symptoms yeast order fml forte 5 ml line, could elevate the plasma tyrosine concentrations (in the early trial from c. In alkaptonuria, the outcome of nitisinone therapy will take many years to consider absolutely, but comprehensive therapeutic examine is justified by the clear relationship between overproduction of a single metabolite and life-shortening tissue manifestations with disabling joint disease. Neurotransmitter ailments and related problems Monogenic defects of neurotransmission have turn into acknowledged as a reason for early-onset, extreme, progressive, and infrequently treatable encephalopathies. Determinations of metabolites in blood or urine are neither sensitive nor specific. In distinction to inborn errors in catabolic pathways, neurotransmitter defects are decided by the Box 12. Even borderline abnormalities could be diagnostic and their recognition requires a strictly standardized sampling protocol and adequate agerelated reference values. Disorders of monoamine metabolism Defects within the metabolism of the biogenic monoamines affect serotonin and/or catecholamine (dopamine and noradrenaline) metabolism. A severe epileptic encephalopathy and progressive studying difficulties could additionally be current. Tyrosine hydroxylase deficiency Tyrosine hydroxylase catalyses the hydroxylation of l-tyrosine to l-dopa, the rate-limiting step in the biosynthesis of the catecholamines dopamine, noradrenaline, and adrenaline. Tyrosine hydroxylase is expressed only in catecholaminergic neurons and the adrenal medulla. Tyrosine hydroxylase deficiency has become included into ideas and classifications of dystonias as the cause of recessive l-dopa-responsive dystonia, but can even present as l-dopanonresponsive dystonia or progressive early-onset encephalopathy. Clinical presentation Clinical signs typically develop between 3 and 7months of age. Most sufferers present a considerable medical improvement already on low doses of l-dopa together with the decarboxylase inhibitor carbidopa, although in contrast to l-dopa-responsive dystonia because of haploinsufficiency of guanosine triphosphate cyclohydrolase I, usually neither the neurological status nor the catecholamine levels in cerebrospinal fluid may be utterly normalized in most sufferers. At the extreme end of the spectrum, nearly no actions are noticed, not even dystonic movements. Some sufferers are more severely affected and present with a progressive neurometabolic dysfunction from early infancy with a progressive infantile encephalopathy characterized by irregular extrapyramidal movements and affecting several cerebral and probably cerebellar methods. It is essential to stress that such patients additionally present signs of significant catecholamine deficiency, corresponding to hypoglycaemia and insufficient stress responses. There is an apparent tendency to preterm delivery with troublesome cardiorespiratory perinatal adaptation. Most infants with tyrosine hydroxylase deficiency develop surprisingly usually till an arrest of motor growth with a characteristic mixture of neurological signs later in infancy. Hypokinesia, marked truncal hypotonia, a mask face, oculogyric crises, myoclonic jerks, and an extrapyramidal tremor can progressively develop. Oculogyric crises are present however, as with the miosis, may go undiagnosed due to prominent ptosis. It appears likely that life expectancy is significantly decreased; (dystonic) cerebral palsy is a probable descriptive (mis-)diagnosis. A characteristic metabolite constellation is found: low concentrations of metabolites of dopaminergic neurotransmission homovanillic acid and 3-methoxy4-hydroxyphenylethyleneglycol within the presence of regular concentrations of metabolites belonging to the serotonin neurotransmission system such as 5-hydroxyindoleacetic acid. Urinary determinations of catecholamines and homovanillic acid turned out to be inconclusive in a number of affected individuals. Treatment and outcome Therapeutic interventions with l-dopa along with the decarboxylase inhibitor carbidopa and selegiline have been in a place to enhance and/or even normalize the clinical image in most sufferers but not all. Treatment with l-dopa has to be began slowly and punctiliously, with doses as little as zero. In such patients, l-dopa can only be increased very slowly, generally over several years. The enzyme is required for the synthesis of each serotonin and the catecholamines. Clinical presentation Clinical symptoms are indistinguishable from those of patients with tyrosine hydroxylase deficiency. About half of the patients current with feeding difficulties, autonomic dysfunction, and hypotonia within the neonatal interval. In the primary few months of life dystonia or intermittent limb spasticity, axial and truncal hypotonia, excessive irritability, oculogyric crises, and psychomotor retardation turn out to be apparent. More mildly affected patients may initially develop unremarkably or solely barely delayed and present with motor retardation, hypokinesia, rigidity, and truncal hypotonia from early childhood. Diagnosis the enzyme deficiency results in accumulation of 3O-methyldopa, 5-hydroxytryptophan, and l-dopa together with low concentrations of the end products homovanillic acid and 5hydroxyindoleacetic acid. Confirmation of the prognosis is by enzyme assay in plasma and finally by mutation evaluation. Treatment and outcome Different approaches using dopamine agonists (pergolide, pramipexole, bromocriptine, and ropinirole) and/or nonselective monoamine oxidase inhibitors (tranylcypromine, phenelzine) have been tried. Response to treatment is variable but consequence appears to be higher in more mildly affected and later-presenting patients. About half of the patients improve on individual treatment regimens and purchase different levels of motor and psychosocial abilities. Dopamine -hydroxylase deficiency Clinical presentation Recessively inherited mutations within the dopamine -hydroxylase gene result in lowered ranges of noradrenaline within central and autonomic noradrenergic neurons. The disorder is characterised by sympathetic noradrenergic denervation and adrenomedullary failure. Syndromes become apparent in adolescence with noradrenergic failure, severe orthostatic hypotension, and ptosis of the eyelids. During childhood fatigue, episodes of fainting, syncopes, and exercise intolerance are typically present. Diagnosis Dopamine -hydroxylase deficiency is classified as a primary autonomic neuropathy. Conditions that result in continual failure of the autonomic nervous system are, therefore, the first differential analysis. Biochemically, dopamine -hydroxylase deficiency is completely different from other circumstances with orthostatic hypotension or autonomic dysfunction. Failure to produce noradrenaline and the ensuing lack of end-product inhibition of tyrosine hydroxylase results in a noradrenaline/dopamine ratio of less than 0. An increase in blood strain and correction of the orthostatic hypotension in response to dihydroxyphenylserine can additionally be diagnostic. Treatment and outcome Dopamine -hydroxylase deficiency is handled with dihydroxyphenylserine. This compound is decarboxylated by l-amino acid decarboxylase to form noradrenaline. Administration of 250 to 500mg twice daily results in an increase in blood strain and sustained aid of the orthostatic symptoms. Without appropriate remedy postural hypotension can result in important accidents or even dying. Later, an analogous therapeutic response was described in the identical scientific constellation for folinic acid.
5 ml fml forte cheap with mastercard
Onset of symptoms is in the first months of life with hypotonia; sometimes affected newborns have difficulties in postnatal adaptation allergy forecast louisville ky fml forte 5 ml discount with amex. Signs of autonomic dysfunction include hypersalivation allergy treatment sublingual immunotherapy 5 ml fml forte cheap overnight delivery, temperature instability, lethargy, hypersomnolence, and episodes of sweating and pallor. Differential diagnosis requires the evaluation of pterins in urine or from Guthrie cards as properly as the dedication of enzyme activity of dihydropteridine reductase in dried blood spots. Urinary biopterin and neopterin values are low within the guanosine triphosphate cyclohydrolase deficiency, whereas 6pyruvoyltetrahydrobiopterin synthase deficiency has excessive neopterin values and low biopterin values. In sufferers with dihydropteridine reductase deficiency, neopterin is regular or barely elevated and biopterin very high. After the biochemical diagnosis, all defects ought to be ascertained enzymatically and, if obtainable, by mutation analysis. Following a analysis of a defect of biopterin metabolism, a lumbar puncture becomes necessary for analysis of the neurotransmitter metabolites 5-hydroxyindoleacetic acid and homovanillic acid in addition to neopterin, biopterin, and 5-methyltetrahydrofolic acid. In sufferers with suggestive encephalopathies and normal phenylalanine values, evaluation of neurotransmitters in cerebrospinal fluid is the only way of diagnosis. Deficiency of neurotransmitters requires treatment with the neurotransmitter precursors l-dopa (3�15mg/kg per day) and 5-hydroxytryptophan (2�9mg/kg per day) in combination with carbidopa (10 or 25% of l-dopa). Patients with dihydropteridine reductase deficiency, as well as, need administration of folinic acid to restore regular cerebrospinal fluid folate concentrations. Normal long-term psychomotor growth may be achieved however outcome strongly is decided by the age when the diagnosis is made and the way rigidly therapy is adopted, particularly in youth. Presentation in kids normally happens within the first decade of life with a mean age of onset of signs being about 7years (range 16months to 13years). The first symptom is normally postural dystonia of one leg with progression to all limbs followed by action dystonia and hand tremor within the next 10 to 15years, during which era cognition remains intact. The dystonia is regularly asymmetrical and accompanied by decreased facial expression or slowing of fantastic finger actions. Diurnal fluctuation is commonly present, with symptoms bettering after nighttime sleep or bed relaxation. Diagnosis In classic circumstances with prominent dystonia of the lower limbs, marked diurnal variation, in addition to worsening of the signs after train, the clinical prognosis of the deficiency is definitely made, specifically in the presence of dramatic and sustained response to l-dopa. Confirmation of the prognosis may be achieved by enzyme analysis in cultured skin fibroblasts or by mutation evaluation. Treatment and end result Treatment relies on l-dopa in combination with 10 to 25% carbidopa. Amounts administered have diversified between 3 and 10mg/kg per day divided into one to 4 doses with the effectiveness of treatment being monitored by the scientific outcome. Tyrosinaemias the steps in tyrosine metabolism starting with the rate-limiting step-the conversion to p-hydroxyphenylpyruvic acid by tyrosine aminotransferase-are outlined in. Intermediates of this tyrosine metabolism are used for manufacturing of catecholamines, dopamine, and the principal pigment of hair and pores and skin, melanin. Tyrosinaemia type I (fumarylacetoacetase deficiency) Clinical presentation Tyrosinaemia sort I is also recognized as hepatorenal tyrosinosis. About one-third of sufferers current acutely in the early weeks of life with failure to thrive, vomiting, hepatomegaly, fever, oedema, and epistaxis; by the end of the first year of life 90% have developed signs. The disease can progress rapidly and dying from hepatic failure typically happens in infancy. The most serious complication is hepatocellular carcinoma which develops in early childhood in one-third of untreated patients. Succinylacetone, shaped from fumarylacetoacetate, is the most particular diagnostic metabolite. Plasma tyrosine values could additionally be regular, resulting in inadequate specificity of this parameter for newborn screening. The danger of hepatocellular carcinoma stays and early liver transplantation was the treatment of selection till nitisinone (2-(2-nitro-4-trifluoromethylbenzoyl)1�3-cyclohexanedione) was launched by Lindstedt and colleagues in 1991. Treatment with nitisinone ought to start as quickly as the analysis is made with a dose of 1mg/ kg per day. Patients must be handled with a diet low in phenylalanine and tyrosine simultaneously introducing nitisinone. The long-term results of nitisinone remedy are encouraging with tremendously reduced incidence of liver injury and hepatic carcinoma. Skin lesions may begin after the attention lesions with blistering, painful palms and soles, and hyperkeratosis. Learning difficulties are an inconstant function in about 50% of patients, however language defects could also be more common with potential impaired coordination and self-mutilation. Plasma tyrosine values attain 20times regular (normal 40�100�mol/ litre) in younger sufferers and 10times normal in others. The scientific features and amino acid analyses are usually adequate for diagnosis, which can be confirmed either by measuring the enzyme exercise in liver or by molecular genetic studies. Treatment and end result A low-tyrosine, low-phenylalanine diet has been used to produce rapid improvement of pores and skin and eye manifestations. The degree of dietary management needed to sustain medical improvement is uncertain. It could also be related to learning difficulties and possibly other neurological complications. Alkaptonuria Clinical presentation In 1902, alkaptonuria was the primary dysfunction to be acknowledged as an inborn error of metabolism by Garrod. It is caused by a deficiency of homogentisate dioxygenase ensuing within the accumulation of homogentisic acid and its oxidized by-product benzoquinone acetic acid. The latter can then be polymerized to form a dark pigment which is deposited in connective tissue. The disorder is extraordinarily uncommon in most populations but occurs with significantly increased frequency in the Dominican Republic and in Slovakia. It is normally normal when handed but darkens on standing (more quickly at alkaline pH) to deep brown or nearly black. Back pain begins within the second and third decade with increasing stiffness because of intervertebral disc degeneration. Greyish discoloration of cartilage is seen within the pinna, and pigment is deposited within the sclera. Abnormal pigmentation is seen within the heart valves and joint cartilages, and pigmented stones are widespread in the prostate. Pigment deposition with involvement of the fibrolipid components of atherosclerotic plaques trigger calcific stenosis of the aortic valve. In 58 sufferers studied by Phornphutkul and colleagues (2002), life-table evaluation confirmed that joint substitute occurred at a mean age of 55years, renal stones at 64years, and cardiac valve involvement at 54years; coronary calcification occurred at a mean age of 59years. Treatment and consequence So far no therapy has been shown to stop the long-term issues.
Order fml forte 5 ml visa
This visual loss is commonly described as a shade coming down or up over a part of or the complete visible subject zopiclone allergy symptoms cheap fml forte 5 ml amex. The onset may be sudden or over a number of seconds allergy symptoms heart palpitations fml forte 5 ml on-line, and typically lasts a couple of minutes earlier than clearing. Even in the absence of neurological signs or pain, these sufferers should bear urgent cardiovascular analysis for ipsilateral carotid artery or cardiac illness, and an erythrocyte sedimentation price and C-reactive protein ought to be obtained. Symptoms suggestive of temporal arteritis, together with headache, scalp tenderness, jaw claudication, muscle fatigue, fever, and weight loss, in sufferers greater than 50 years of age, ought to immediate instant corticosteroid therapy and temporal artery biopsy. These transient visible obscurations are often a symptom of underlying disc edema of any cause, most commonly papilledema. Table 2 Causes of transient monocular visual loss or blurriness Vascular Transient ophthalmic artery occlusion Transient central retinal artery or branch retinal artery occlusion Ischemic optic neuropathy (from vasculitis) Arteriovenous malformation (steal phenomenon) Central retinal vein occlusion Transient visible obscurations (optic nerve head swelling or drusen) Orbital tumor (gaze-evoked visual loss) Angle closure glaucoma Hyphema (blood in the anterior chamber) Dry eyes Corneal edema Table 3 Causes of monocular visual loss (permanent or transient) associated with pain Vascular visible loss from internal carotid artery dissection Carotid occlusion Ocular ischemic syndrome Giant cell arteritis Angle closure glaucoma Ocular trauma More persistent monocular visible loss is characterised greatest by the tempo of onset and the age of the affected person. Central monocular visible loss in the young grownup over days or weeks most likely represents optic neuritis. Sudden monocular visible loss within the older affected person doubtless reflects vascular disease. The former is generally thought-about a disease of small vessels (associated with hypertension and diabetes) occurring in sufferers with congenitally small, crowded optic nerve heads. The latter may end up from emboli from the great vessels (including the internal carotid artery) or the heart. Compressive lesions of the optic nerve end in gradual monocular lack of central vision or visible subject. There could additionally be complaints of headache, periocular ache or numbness, proptosis, diplopia, ptosis, or change in pupillary look, suggesting orbital or superior orbital fissure involvement. Endocrine dysfunction might replicate an underlying pituitary tumor or a craniopharyngioma. Other causes of transient binocular visible loss embrace the transient visible obscurations of bilateral disc edema, focal seizures involving the occipital lobes, and transient ischemic attacks, especially throughout the posterior cerebral circulation. Head trauma, especially in children, may lead to cortical blindness that lasts hours to days. Permanent binocular visual loss can result from injury to each optic nerves, the chiasm, or the retrochiasmal visual pathways. When compressive in etiology, bilateral optic neuropathies could reflect bilateral orbital lesions or, extra commonly, lots within the region of the chiasm. As long as central visual acuity is spared, patients might not notice visible area defects suggestive of chiasmal and retrochiasmal lesions. In addition, patients with visual area defects could complain of associated neurological symptoms similar to headache, hemiparesis or numbness, and aphasia, depending on the anatomic location of the lesion. Most chiasmal and retrochiasmal visual loss is a result of cerebral mass lesions and strokes. Oscillopsia Visual field check Reliable Normal in each eyes Abnormal Not dependable -Repeat another day -Change technique -Use confrontation One eye irregular Two eyes irregular Disease involving the attention, retina, or optic nerve No Does the deficit respect the vertical meridian (at least partially) It has a broad differential prognosis, including vascular, neurological and ophthalmic causes (Table 1). The time period amaurosis fugax is commonly used to describe transient monocular visual loss, but this term has turn into synonymous with transient monocular visual loss of ischemic etiology The time period transient monocular blindness is also usually used to describe transient monocular visual loss, but implies complete lack of imaginative and prescient. Homonymous visual loss is commonly mistaken for monocular visible loss on the facet with the temporal defect and, thus, it is necessary to ask whether or not visual loss was famous within the different eye when the affected eye was coated through the episode. The presence of a precipitating factor may help to slender the differential prognosis. Although nonspecific, the pattern of visible loss can help to slim the differential diagnosis. A history of polymyalgia rheumatica, connective tissue disease, hematologic abnormality The visual loss is usually complete, but generally only a sector of the visual field is concerned. The duration of visible loss is minutes, however often lower than 15 min, following which vision spontaneously returns to baseline. Rarer causes of retinal emboli embody common carotid artery atheromatous plaque, atrial myxoma, and paradoxical emboli touring through a cardiac septal defect or pulmonary shunt. Echocardiography, especially transesophageal echocardiography, is required to identify structural cardiac abnormalities related to intramural thrombus formation and paradoxical embolism, and can be used to assess the aortic arch for atheromatous plaque. Prolonged electrocardiographic monitoring could also be required to diagnose paroxysmal arrhythmias. Carotid stenting has been proposed as an various to endarterectomy, however has been associated with a better threat of periprocedural stroke. Ophthalmic Artery Stenosis and Occlusion Ophthalmic artery stenosis and occlusion are reported as rare causes of transient monocular visual loss. Transcranial Doppler ultrasound and formal cerebral angiography are sometimes needed to affirm the analysis. Vascular Causes Retinal Emboli Retinal emboli are a standard cause of transient monocular visual loss. The affected person usually reviews a sudden onset of Carotid Artery Stenosis and Occlusion Transient monocular visual loss could be attributable to stenosis or occlusion of the inner or frequent carotid artery. It is commonly precipitated by exposure to bright gentle, but can even occur after meals, postural adjustments, or sexual activity. In sufferers with a cardiac source of embolism, anticoagulation with warfarin and remedy of the underlying cardiac illness is indicated. Patients with atherothrombotic illness ought to be commenced on antiplatelet therapy. Carotid stenting could be considered in high-risk surgical candidates, however is associated with a better danger of periprocedural stroke. The episodes are often precipitated by postural changes and are thought to arise because of ischemia of the afferent visible system. Those with no historical past of migraine require neuroimaging to exclude a structural lesion, even when they describe a typical aura and have an unremarkable examination. Similar attacks of transient monocular visual loss could be brought on by retinal vasospasm. Treatment for migraine includes avoidance of precipitating elements and use of abortive treatments at the onset of assaults. Patients with frequent or disabling migraines often profit from prophylactic therapies, such as propranolol, amitriptyline, or topiramate. Episodes of transient monocular visual loss due to vasospasm typically remit with calcium channel blockers. Occipital Seizures Occipital seizures typically produce binocular elementary optimistic visible phenomena with related visible loss. In distinction with migraine, the positive visible phenomena include a quantity of, brightly colored, circular figures in the contralateral hemifield. The optimistic visible phenomena usually final for lower than a minute, however the vision loss can persist for a quantity of days postictally. The seizures can usually be managed medically, though surgical excision of the epileptogenic focus may be required in refractory instances. Neurological Causes Migraine Aura Migraine aura is a typical reason for episodic transient homonymous binocular visible loss.
Fml forte 5 ml purchase line
Because these lesions grow slowly allergy shots and weight loss fml forte 5 ml buy with amex, recurrences have been detected even more than a decade after complete resection allergy symptoms to milk fml forte 5 ml discount mastercard. For that reason, any progression of signs after surgical procedure ought to prompt an analysis for recurrence. When full resection is infeasible, some evidence means that adjuvant radiation therapy may be of benefit. Spinal Meningiomas Spinal meningiomas are typically benign, intradural-extramedullary lesions that arise from the arachnoid cap cells. These lesions trigger neurological symptoms by compressing the spinal wire from the extramedullary compartment. Meningiomas are the second commonest intradural-extramedullary tumors, behind nerve sheath tumors. Complete resection is usually feasible, as a end result of a surgical aircraft between the lesion and the spinal twine is usually apparent. Nerve Sheath Tumors: Schwannomas and Neurofibromas Nerve sheath tumors include schwannomas and neurofibromas. These lesions are usually intradural-extramedullary, though they may have an extradural element. Both tumor sorts are believed to come up from Schwann cells, though schwannomas and neurofibromas have distinct histological, organic, and surgical features. Neurofibromas typically contain diffuse enlargement of the affected nerve, making surgical separation of the lesion and nerve tougher. Neurofibromas may occur sporadically or in association with neurofibromatosis, a genetic condition Myxopapillary Ependymomas Myxopapillary ependymomas are rare tumors and distinct from intramedullary ependymomas. Complete resection of those lesions is the treatment of selection and, if neurological operate is maintained after surgery, is associated with long-term preservation of operate. The recurrence fee for these lesions is 280 Spinal Cord Tumors, Treatment of during which a number of neurofibromas may be found. Schwannomas sometimes develop eccentrically from the nerve, usually enabling the identification of a surgical dissection plane between the tumor and nerve. The optimal treatment for both symptomatic neurofibromas and schwannomas is surgical resection, with preservation of as a lot functional nerve as potential. Malignant nerve sheath tumors are related to excessive charges of recurrence and a poor total prognosis. Thoracic Spine Stabilization Further Reading Balmaceda C (2000) Chemotherapy for intramedullary spinal twine tumors. More than a century ago, Werdnig and Hoffmann independently described a dysfunction with progressive weakness that began in infancy, resulted in death at an early however inconstant age, and was characterised pathologically by anterior horn cell loss. Kugelberg and Welander later described a far milder, slowly progressive motor neuronopathy marked by chronic neurogenic adjustments and muscle weakness affecting proximal muscle tissue. The previous decade has witnessed dramatic advances in molecular diagnostics and the range, availability, and effectiveness of medical management. There can also be immense hope that efficient remedies for this disease shall be developed within the near future. Unfortunately, this correlation is simply too inaccurate to successfully guide prognosis for any individual subject. Within every illness classification there are relatively weak and powerful sufferers with potentially vastly totally different medical care wants, disease morbidity, and prognosis for survival. Traditionally, these infants suffer from early respiratory dysfunction and progressive malnutrition. Historically, with the restricted support out there, few survived to their second birthday. With trendy medical care, most survive previous 2 years of age, with many surviving well into their second decade. These youngsters often develop signs between 6 and 12 months of age, although many develop initial symptoms of weak spot and lowered tone earlier than 6 months of age. This mutation is each frequent and panethnic, with a carrier frequency of between 1 in 40 and 1 in 50. The presence of this thymidine nucleotide seems to create a more viable exon splice website in exon 7. This residual protein is enough to maintain fetal viability however insufficient typically to keep away from the development of scientific symptoms. Areflexia is nearly universal, although some could retain deep tendon reflexes briefly early in the disease course. Earliest signs develop in the first few weeks of life, although often the symptoms are underappreciated by mother and father and pediatricians for weeks to months till the degree of weak point and low tone turns into impossible to ignore. A paradoxical respiration sample, marked by chest wall incursion and stomach expansion throughout inspiration (resulting from intracostal muscle weak point and relative preservation of diaphragmatic function) is commonly seen and suggests ongoing pulmonary compromise and the need for intervention to help ventilation. Swallowing dysfunction, choking, and aspiration are almost common in the months following symptom onset. By definition they develop the power to sit however fail to develop the power to walk independently. Electromyography is not routinely carried out, save as part of an analysis for kids with unusual shows or where genetic testing is adverse or inconclusive. Muscle biopsy demonstrates grouping of type 1 and a pair of muscle fibers versus the traditional checkerboard sample, in preserving with a neurogenic disease. In the early stages of the illness, the biopsy might present universally small fibers, with type 1 fibers being smaller than sort 2, making the histological diagnosis tough. Cerebral pathology should be suspected if alertness is diminished or infantile reflexes persist. Spinal Muscular Atrophy 283 Spinal twine disorders, including trauma of the cervical spinal wire, ought to be thought-about as supported by clinical context. Congenital myopathies, including centronuclear myopathy, nemaline myopathy, central core disease, congenital muscular dystrophy and (particularly) myotonic muscular dystrophy could current with floppiness at delivery. Congenital myopathies may also present with a clumsy gait and issue walking up and down stairs, although onset from early infancy usually distinguishes these groups of diseases. Muscle biopsy and electrophysiological examination ought to lead to the appropriate analysis in unclear medical instances. Prevention the identification of the causative gene has dramatically advanced the function of genetic counseling. Although universally accepted follow has not been fully established, expectant moms routinely receive service testing early in pregnancy or, ideally, earlier than turning into pregnant. If both dad and mom are identified carriers, chorionic villus sampling or amniocentesis could be carried out to establish affected fetuses. Pulmonary toileting techniques, including guide chest physiotherapy, mechanical suctioning and use of cough assist gadgets are crucial to keep away from pneumonia related to aspiration or sickness. Although tracheostomy stays an possibility for affected children, noninvasive air flow is increasingly most popular by many mother and father and practitioners within the subject. Intensive long-term physical remedy providers ought to be maintained for all affected individuals. Support units including bracing, switch devices, mechanical standers, and, particularly, motorized wheelchairs should be aggressively procured in an effort to maximize independent operate.
Red Date (Jujube). FML Forte.
- How does Jujube work?
- Dosing considerations for Jujube.
- Liver disease, muscular conditions, ulcers, dry skin, wounds, diarrhea, fatigue, and other conditions.
- What is Jujube?
- Are there safety concerns?
Source: http://www.rxlist.com/script/main/art.asp?articlekey=96108

Cheap fml forte 5 ml
This was invasive and technically tough allergy medicine anxiety generic fml forte 5 ml visa, and has to a large extent been outmoded by new methods allergy treatment muscle testing 5 ml fml forte generic with mastercard. In infants with suggestive signs, biochemical profiling, with measurements of glucose, lactate, and ketones, can recommend the correct diagnosis. If frozen tissue has been saved, enzymology Glycogen storage disease sort V (McArdle disease) Biochemistry McArdle described a patient who suffered exercise-induced myalgia in whom lactate fell during ischaemic train rather than rising, suggesting a defect in glycogenolysis. Patients are asymptomatic during low depth, aerobic train, when muscle depends on fatty acid oxidation for energy, however during anaerobic train patients rely on glycolysis and develop signs. Electron microscopy can also be helpful if structurally irregular glycogen is present. A diagnostic fast, with measurement of glucose, lactate, and ketones also can present useful info. This check is all the time disagreeable for the patient, and might precipitate rhabdomyolysis, and is now seldom performed. A number of the enzymes involved could be assayed in leucocytes, however for others muscle is needed (Table 12. Testing for particular person enzymes is laborious and costly and, more and more, molecular genetics is changing into the diagnostic test of selection. Diagnosis and administration of glycogen storage disease type I: a follow guideline of the American College of Medical Genetics and Genomics. Adult polyglucosan physique disease: pure history and key magnetic resonance imaging findings. Pharmacological and dietary therapy for McArdle illness (glycogen storage illness kind V). Autophagy and mitochondria in Pompe illness: nothing is so new as what has long been forgotten. After rapid carrier-mediated absorption throughout the intestinal epithelium, fructose is metabolized (mainly within the liver) by the enzymes ketohexokinase (fructokinase), aldolase B, and triokinase, eventually being converted into glucose or glycogen. Dietary sugars-burgeoning constituents in meals and drinks worldwide- have undesirable effects on these with restricted capability to metabolize fructose, together with severe illness or dying in younger sufferers. Three inborn errors of fructose metabolism are acknowledged and these disorders are vivid examples of gene�environment interactions: 1. Essential or benign fructosuria because of fructokinase deficiency-a very rare disorder with apparently no unwell effects. Hereditary fructose intolerance (fructosaemia)-an autosomal recessive illness caused by deficiency of aldolase B. May come to mild at any age with postprandial belly pain and vomiting, symptomatic hypoglycaemia (which may induce seizures), hypophosphataemia, acidosis, and other metabolic disturbances after consumption of offending foods and drinks. Unrecognized illness causes failure to thrive/growth retardation, a Fanconi-like renal syndrome with nephrocalcinosis, and jaundice with deadly liver harm. Parenteral infusion of fructose or its congeners might trigger dying from acute hepatorenal damage. Diagnosis formerly depended on a controlled intravenous fructose challenge take a look at or demonstration of poor aldolase B isozyme exercise in liver or small intestinal biopsy material; currently molecular analysis of the aldolase B gene is most well-liked and usually decisive. Treatment requires establishment of a strict sugar-exclusion food plan supplemented by folic acid and vitamin C. Early analysis and dietary modification are critical for well-being and regular development. Fructose-1,6-diphosphatase deficiency-a very rare illness of infancy and childhood associated with failure of hepatic gluconeogenesis inflicting bouts of extreme hypoglycaemia, ketosis, and lactic acidosis provoked by infection and starvation. Metabolic decompensation is provoked by dietary fructose, associated sugars, and/or ketogenic fats. Diagnosis is decided by enzymatic assay of fructose 1,6-diphosphatase in fresh liver biopsy samples or molecular analysis of the cognate gene. Treatment requires a fructose-exclusion food regimen containing ample carbohydrate vitality, restricted fat, and protein. Acute episodes of acidosis or hypoglycaemia are managed by intravenous glucose infusions, with bicarbonate if required. Given the primacy of fructose and sucrose in the phenotypic expression of those disorders, an understanding of the driving forces answerable for the disease is of central significance for holistic medical management. Certainly, the comparatively rare conditions which are as a outcome of disturbance of fructose metabolism share features with environmentally pushed syndromes of obesity, nonalcoholic hepatic steatosis, and hyperlipidaemia. In the background, a succession of regrettable historical events led to the introduction of sugar primarily into the modern European food regimen. Insatiable calls for for sugar obtained in plantations by foreign slave labour in New World colonies has left many painful penalties, for example, the extensive geographic dissemination of sickle cell illness and different effects such as financial deprivation and social injustice. Consumption of fructose and sucrose the sugar commerce expanded in the course of the 16th to nineteenth centuries ce because the mass seize and buying and selling of slaves took hold. Partially modelled on earlier Arabic trading in Africa, the usage of slave labour in the sugar plantations in New World and different colonies has a haunting legacy. Nowadays, however, this history has an ironic payback within the consequential results of sugar on global well being. The mass industrialization of sugar production declares the nature of the human appetite for sweet flavours, and exogenous sugar is added to food and drinks in fashionable societies across the world. Disorders of fructose metabolism and its assimilation mirror the ubiquity of sucrose and fructose in the food plan. Most individuals in developed countries ingest 50 to 100 g fructose equivalents daily within the food plan. Global manufacturing of sugar (sucrose from cane and beet) continues to rise, but over the past three many years, novel manufacture of fructose as a high-fructose corn syrup, enzymatically derived from starch in maize (corn), has additionally burgeoned. This intensely sweet sugar, first enzymatically produced commercially from extra maize within the early Nineteen Seventies, primarily from United States mid-Western agriculture, has been popular in the United States of America, Mexico, and China, however is now manufactured and used within the European Union and Australia. Introduction Fructose is a vital and burgeoning element of the modern food plan; it happens as a free monosaccharide in fruit, nuts, honey, and a few greens however is now abundant and practically ubiquitous in well-liked, mass-produced drinks. Free fructose is launched from the disaccharide sucrose within the gut lumen by the sucrase�isomaltase advanced at the brush-border membrane of the mucosal epithelium; in fashionable times, sucrose is the principal supply of fructose for most persons. Finally, the sugar alcohol, sorbitol (a constituent of medicines and tablets, as properly as diabetics), is transformed quantitatively to fructose within the liver and intestine. Inborn errors of fructose metabolism present distinctive examples of gene�environment interactions in current economic history. Emergence of those situations displays revolutionary dietary modifications resulting from the mass industrialization of sugar farming and manufacture. Until lately, all largescale sugar manufacturing came from sugar beet and sugar cane, which in fashionable occasions yield merchandise that are indistinguishable in taste and composition. Cane sugar, principally from tropical international locations, represents about 80% of the worldwide market and is usually cheaper to manufacture than beet sugar, which has enjoyed political and economic assist from native farming techniques, generally in nontropical international locations. Direct market competition of beet with cane sugar had occurred in postcolonial instances because beet sugar-producing nations mitigated losses on exports by high income in home markets, thus permitting growth of home beet cultivation. Brazil, one of many first since its early Portuguese colonization, and nonetheless the biggest producer, controls half the global market but pays subsidies measured in billions annually to its sugar industry. Until very just lately, a complex tariff fee system offering direct help of domestic sugar production was used in the United States of America, which maintained the worth there as much as 90% larger than the world market worth at an annual charge of 3.
Cheap fml forte 5 ml with mastercard
Treatments Depending on the underlying drawback affecting style allergy vinyl symptoms fml forte 5 ml order, there are some therapeutic methods obtainable allergy treatment desensitization buy generic fml forte 5 ml on line. For instance, infections and inflammation of the mouth surfaces and perioral buildings could be treated with antibiotic or anti-inflammatory drugs. Diminished style operate associated with smoking or poor oral hygiene could be reversed, to some extent, by cessation of smoking and improved hygienic measures. This marked a significant milestone in the early development of neurology as a specialty as neurologists sought to develop or undertake instrumentation that might distinguish the specialty and assist remark, diagnosis, and remedy. Taylor acquired his medical diploma from the University of Pennsylvania in 1878, and then practiced pediatrics, neurology, and physical medicine in Philadelphia. Early variations had an open loop deal with, but some versions after 1920 had a solid handle with a pointed tip for eliciting cutaneous reflexes. The special feature of this hammer is that the shape of the putting floor is like the outer surface of the extended hand, palm downward, which is most often used in acquiring tendon jerk. The rounded apex finish is adopted (sic) to attain the biceps tendon at the bend of the arm (and, in accordance with S. Weir Mitchell in his clinical apply on the Philadelphia Infirmary for Nervous Diseases. Mitchell was routinely photographed interviewing or examining patients while holding a Taylor hammer in his proper hand. Early variations had an open loop deal with, but some variations after about 1920 had a strong handle with a pointed tip for eliciting cutaneous reflexes. Decades later, the American Academy of Neurology included the Taylor hammer into its original emblem (which has since been outdated by another design). This text was later translated into Italian (Manuale Delle Malattie Dei Bambini, 1905). In addition to his academic accomplishments, Taylor was highly regarded as a clinician. The illness was first reported in 1881 by Tay, a British ophthalmologist who described a 1-year-old child with psychological retardation and a cherry-red spot within the retina, and in 1887 by Sachs, an American neurologist who famous the central nervous system pathology and recognized the elevated prevalence of the illness amongst Jews. In 1970, a 12 months after the enzyme defect was recognized, screening applications had been begun to detect carriers (heterozygotes) and provide genetic counseling on family planning. Current pointers recommend heterozygote screening even when solely one of the mother and father is from Eastern European Jewish heritage. The base substitution in intron 12, seen in Jewish carriers, has not been found in the affected non-Jewish population. It was not until the 1930s that the chemical foundation for the illness was discovered. These complicated lipids, that are common components of the neuronal plasma membrane, are composed of sphingosine, fatty acids, and a hydrophilic oligosaccharide chain. On ultrastructural research, the excess storage materials produces a concentric, lamellar look within the lysosomes referred to as membranous cytoplasmic bodies. In the retina, this buildup of lipids within the ganglion cells within the parafoveal area produces a hazy whitish ring, making the traditional reddish look of the macula standout much more, hence the cherry-red spot. Knockout mice with disrupted ganglioside synthesis, however, have only minor neurological deficits. The first signs may be an extreme startle reaction to loud noises or failure to achieve regular developmental milestones. Infants are hypotonic, unable to sit alone, and sometimes fail to develop any language. From this level onward, children decline into an more and more extra vegetative state. Progressive macrocephaly occurs because of the increased amount of intracellular storage material, although the liver and spleen are regular in size. Developmental regression follows, with spasticity and seizures widespread by the end of the primary decade. Loss of imaginative and prescient happens later than in the infantile disease and is clear by optic atrophy and retinitis pigmentosa. Because accumulation of the gangliosides begins in utero and is already properly developed by the second trimester, postnatal enzyme replacement or bone marrow transplantation are unlikely to be effective. A variety of these buildings, together with areas outside of the temporal lobe, are considered to comprise an interconnected circuit referred to as the limbic system, which is crucial for motivation, studying, and emotion. The hippocampus and amygdala (so named due to their anatomical resemblance to a seahorse and an almond, respectively) are the most wellstudied components of this method. As mentioned in additional element later, the hippocampus performs a crucial position in human reminiscence, whereas the amygdala seems to be responsible for many features of motivation and emotional processing. Another notable region inside the temporal lobe is primary auditory cortex, which is located inside the Sylvian fissure. Studies of patients with mind damage or neurological illness affecting the temporal lobe have contributed much to our understanding of the role that the area might play in cognition. In recent years this has been complemented by evidence from useful imaging methods that allow exploration of patterns of temporal lobe activity associated with cognitive processing in healthy individuals. The diagram also reveals different regions extensively connected to the medial temporal lobe, such because the fornix, basal forebrain, thalamus, and prefrontal cortex. This imaginary boundary is approximately coextensive with the dorsal and posterior border of Brodmann space 22, on the superior temporal gyrus, and Brodmann area 37. On the inferior floor, the posterior extent is outlined by an imaginary line working from the preoccipital notch to the anterior termination of the calcarine sulcus. This form of surgical intervention is usually an efficient therapy for seizures of temporal lobe origin that are proof against drug therapy: it has been demonstrated to produce a marked reduction in seizure frequency or even full abolition of seizures. Excisions are discrete and focal, often involving temporal lobe constructions in a single hemisphere solely, thereby allowing researchers to examine the different roles of the left and right temporal lobe. It should also be noted that some temporal lobe structures, particularly the hippocampus, parahippocampal gyrus, and uncus, are particularly susceptible to brain harm, permitting researchers to investigate the differential involvement of those regions in cognition. In contrast, sufferers with lesions to the left temporal lobe are poor in verbal reminiscence tests such as remembering the major points of a narrative read to them a quick time earlier than. These patterns of practical hemispheric asymmetry are sometimes mirrored in structural anatomical variations that might be observed even in healthy individuals. Such anatomical differences are current from very early in development, maybe linked to prenatal hemispheric gene expression asymmetries appearing as early as 12 weeks of gestation. However, the view that the left hemisphere is devoted to verbal processing and the best to nonverbal aspects of cognition is kind of actually too simplistic. For example, sufferers with damage to the left hemisphere, particularly to the temporal lobe, present a variety of deficits of their capability to produce phrases and, in some cases, to perceive written and spoken language (aphasia). However, lesions to the best hemisphere also end in difficulties with some aspects of linguistic processing, such as understanding the intonation, stress, and rhythm of speech (prosody). These findings point out that the right and left hemispheres are each important for aspects of language.
Purchase 5 ml fml forte with amex
Placement of a everlasting ventriculoperitoneal shunt may be necessary when hydrocephalus complicates tuberculous meningitis allergy forecast akron ohio buy discount fml forte 5 ml online. The prognosis of meningitis is instantly associated to the medical stage of illness when antituberculosis chemotherapy is began new allergy treatment 2012 fml forte 5 ml buy discount online. The majority of patients diagnosed in stage 3 will both die or have extreme neurological sequelae. Some patients recognized in stage 2 have an excellent consequence, whereas others have persistent neurological deficits. In common, the prognosis is worse for infants and the very old, immunocompromised patients, malnourished patients, patients with associated disseminated illness, and sufferers who present with elevated intracranial pressure. Mental retardation and impairment of mind and judgment, dementia, or some extent of habits or studying dysfunction are widespread problems in patients presenting with stage 2 or 3 disease. The most common varieties trigger obesity, hypogonadism, sexual precocity, diabetes insipidus, and growth retardation. In the United States, the major methods of preventing tuberculosis an infection and illness center on the public health actions of contact investigation are figuring out and treating tuberculosis infection. Identifying infected persons and rapidly treating them is the best way to stop additional cases of tuberculosis. Arseni C (1958) Two hundred and one circumstances of intracranial tuberculoma handled surgically. Skin lesions embody hypomelanotic macules, facial angiofibromas, Shagreen patches, and ungual fibromata. Lymphangiomyomatosis of the lung is a uncommon however potentially fatal complication that affects women between the ages of 20 and 40 years. As sudden pneumothorax and chylothorax may happen, screening of asymptomatic girls ought to be considered. Finally, ocular lesions could additionally be of diagnostic utility however are not often symptomatic and embrace hamartomas and achromatic patches. Neurologically, many sufferers will have problem controlling seizure disorders or behavioral issues requiring referral to a specialist. An increased understanding of the genetic reason for the disease and the underlying dysregulation of the mammalian goal of rapamycin pathway has led to medical trials of the mammalian target of rapamycin inhibitors, including sirolimus and everolimus. There seems to be a comparatively excessive rate of mosaicism among founders, which might mitigate the phenotype. Neuropsychiatric analysis is indicated for youngsters with developmental delay, faculty efficiency issues, or a history of behavioral difficulties at residence or in school. When benign as nicely as malignant brain tumors are included, the incidence is sort of 3 times that of malignant brain tumors alone. Survivors additionally may endure severe neurocognitive deficits or late health effects corresponding to second main malignancies because of remedy. In this text, what is known concerning the distribution of main tumors of the brain in human populations is reviewed to provide a greater understanding of the kinds of people most impacted by the disease. Further, the article consists of an overview of the molecular and genetical traits of the illness, and the necessary thing recognized and instructed risk factors for mind tumors from epidemiological research. Some environmental brokers, particularly ionizing radiation, are clearly implicated within the etiology of brain tumors but account for few cases. These tumors are unique because of their location within the bony structure of the cranium. Furthermore, histologically benign tumors could cause related signs and outcomes as malignant tumors as a result of development of tumor tissue inside the cranial area might injury regular tissue. For this reason, cancer registries now embrace both benign and malignant intracranial tumors. For simplicity, this group of tumors might be known as brain tumors or, when benign tumors are excluded, brain cancer. There is usually wide variation within the particular histological forms of tumors included in printed incidence and mortality charges, and whether or not these tumors are reported individually or as grouped data also can differ. In this entry, we review descriptive knowledge that features each benign and malignant instances utilizing knowledge from the Los Angeles County Cancer Surveillance Program. As will become clear from later discussion of etiological research (see section Suggested Causes of Brain Tumors), extra is understood concerning the causes of benign histological sorts corresponding to meningiomas than concerning the etiology of neuroepithelial tumors, that are more frequent than meningiomas and are often malignant. Another challenge when comparing incidence and mortality charges throughout publications relates to whether clinically recognized tumors are included. In common, for comparatively inaccessible most cancers sites, the next price of microscopic affirmation increases the likelihood that a neoplasm truly existed and that it was appropriately classified. Pathological Classification the evaluation of scientific and epidemiological characteristics of brain tumors and the research of genetical or environme ntal threat components requires the accurate classification and histopathological typing of brain tumors. Brain tumors are diverse with respect to morphological, cytogenetical, and molecular 552 Encyclopedia of the Neurological Sciences, Volume four doi:10. Astrocytic tumors which are grades 1 and a couple of are usually categorised as astrocytomas, these which are grade three are categorized as anaplastic astrocytomas, and those which are grade 4 are classified as glioblastomas. Nerve sheath tumors, referred to as neuromas, neurilemmomas, or schwannomas, arise in the Schwann cells of the nerve sheath. These comparisons recommend that brain tumor incidence continues to improve with age throughout life however that rates in the past may have been underreported in the oldest age teams. Therefore, historical comparisons of brain tumor charges from totally different registries are impacted by changes in ascertainment by age over time. For all histological sorts and races combined, the age-adjusted abstract incidence charges of major brain tumors are roughly the identical in women and men. Rates of neuroepithelial tumors are decrease amongst black women and men than among non-Hispanic whites, but are roughly the same for meningiomas among both males and females (Table 2). Rates are lower in Los Angeles Asian adults than in African-American Hispanic, or non-Hispanic whites. Low charges also have been reported in Asian populations in Japan, India, and among the many Chinese in Singapore. Among every racial group, rates are often greater in migrant populations than in native populations that remain in their country of origin. Summary of Descriptive Epidemiology Perhaps, the most important discovering from this evaluation of the descriptive epidemiolsogy of mind tumors is that the pattern of incidence and survival each range significantly by histological type, age, and gender. For males, this development is clear for neuroepithelial tumors and nerve sheath tumors. The exception to that is meningiomas, which shows the inverse relationship amongst each males and females. Suggested Causes of Brain Tumors Ionizing Radiation the occurrence of extra cases of brain tumors after high-dose publicity to ionizing radiation is well-established. Survival charges for all tumors differ considerably by location, conduct, histological type, and age. Additionally, elevated danger of brain tumors has been observed amongst atomic bomb survivors, with greater threat noticed among individuals uncovered in childhood than those exposed in adulthood. The association between low-dose publicity to radiation and brain tumors is extra controversial. Prenatal exposure to diagnostic radiography has been associated to extra pediatric mind tumors in a quantity of studies because the preliminary report in 1958. A recent study observed an affiliation between bitewing examinations at any age, or panorex movies taken at an early age on an yearly foundation and threat of meningioma.
Fml forte 5 ml buy on-line
They ask for assist when the results for household allergy forecast pleasanton ca discount fml forte 5 ml on-line, income allergy west discount 5 ml fml forte with visa, and employment have become too great. Professional remedies are inclined to emphasize a cognitivebehavioral approach with particular concentrate on identifying relapse cues and triggers. Pentazocine (Talwin) and meperidine (Demerol), synthetic opioids, had been each initially launched as nonaddicting analgesics. Today, synthetic opioids such because the fentanyl compounds are lots of of occasions more potent, and subsequently more harmful, than morphine or heroin. Naturally occurring opioids include tincture of opium (Laudanum), camphorated tincture of opium (Paregoric), morphine, and codeine. Semisynthetic opioids embrace hydromorphone (Dilaudid), oxycodone (Percodan), and diacetylmorphine (heroin). Synthetic opioids embrace meperidine (Demerol), methadone (Dolophine), and propoxyphene (Darvon). Incidence and Pattern of Use Heroin is processed from morphine, a naturally occurring substance extracted from the seed pod of certain kinds of poppy vegetation. Historically, new cultivation of opium poppies in South America has elevated the purity and lowered the price of avenue heroin. A massive proportion of latest users smoked, sniffed, or snorted more and more pure and increasingly available heroin, due partially to new manufacturing in South America. Both produce similar syndromes, which can easily be mistaken for paranoid schizophrenia or other psychotic processes. Because sympathetic overactivity is commonly discovered with functional psychoses, one should turn to the historical past and to urine toxicology to assist rule out panic disorder, paranoid schizophrenia, and mania. Heroin (and Other Opioids) History Opium smoking has been practiced for centuries. Indeed, within the seventeenth and eighteenth centuries the British helped develop extra opium addicts in China to find a way to trade Indian opium for Chinese tea and silks. Each new form of opiate treatment has inevitably become a model new kind of addiction downside. They are richly found in the fast interneurons that are thought to modulate neuroactivity, notably inhibitory activity. Neonatal habit: There is a consensus that heroin use is more harmful than methadone, despite the very fact that withdrawal for the neonate on methadone is more difficult. Methadone upkeep quite than methadone withdrawal is recommended for the pregnant addict. Major maternal problems include malnutrition, venereal illness, hepatitis, pulmonary issues, and preeclampsia. Problems of early infancy include potential development retardation, potential delayed motor improvement, and a few behavioral abnormalities. The most sensitive indicators are hand tremor, trapezius electromyography, and coronary heart price. Talwin (pentazocine) is an antagonistic and will trigger withdrawal in a methadone patient. Central a-adrenergic stimulation decreases blood strain, pulse, and cardiac output. The site of action is the locus ceruleus (blue spot), associated with the limbic system. Withdrawal signs are largely as a result of sympathetic overactivity, which is blocked by clonidine. Adverse affects embrace high dose, sedation, hypotension, sluggishness, drowsiness, and dry mouth. Clonidine is contraindicated in coronary insufficiency, myocardial infarction, renal failure, and pregnancy. Clonidine withdrawal symptom consists of rebound hypertension (especially in hypertensives), nervousness, agitation, and headache. Resistant signs embrace insomnia, anxiety, restlessness, and muscular aches and pains. Severe intoxication: Unresponsiveness, coma, bradycardia, hypotension, decreased respiration, hypothermia, and pinpoint pupils. Chronic use: Dose tolerance, improve in social issues, dysphoria, increased somatic issues, motor retardation, and blunting of affect. Overdose: Special issues embrace unknown dosage, blended medicine, adulterants, and medical problems. Signs and signs include stupor, coma, pinpoint pupils, decreased pulse and respiration fee, and pulmonary edema. This treatment maintains dependence in change for social 346 Substance Abuse rehabilitation. Methadone results are the same as opiate effects, corresponding to decreased libido, weight gain, delayed ejaculation, and apathy. Risks embrace overdose, mixed usage (particularly with alcohol), and diversion to street drugs. Opioid antagonists An opiate antagonist will forestall euphoria and dependence in a nonaddict. Dysphoric unwanted side effects include sedation, visual disturbances, racing ideas, and hallucinations. The most compliant patients are medical professionals whose licenses to follow are at stake. Other identifiable causes of sudden toddler demise embrace inherited issues of fatty acid oxidation, cardiac channelopathies, suffocation and infanticide, and infection. Environmental stressors embrace inclined or facet sleep place, soft bedding, mattress sharing, and delicate (respiratory) infections. During the crucial developmental period that occurs within the first 6 months of life, there are speedy adjustments in brainstem function together with autonomic regulation and maturation of cardiorespiratory control, arousal, and sleep state. Environmental stressors, both intraand extrauterine, similar to poverty, exposure to tobacco, alcohol, or illicit medication, might directly impression neurotransmitter techniques critical to homeostatic control within the developing brain. In animal fashions, exposure to nicotine during fetal growth alters the expression of nicotinic acetylcholine receptors liable for autonomic function. In the developing brainstem of human infants, nicotinic acetylcholine and serotonin receptors are intently linked. It is obvious that arousal from sleep triggered by abnormal levels of carbon dioxide and oxygen is important for the initiation of protecting airway responses. Arousal Arousal is a posh process involving the interplay of subcortical (brainstem) and cortical mind constructions and the suggestions loops between them. The brainstem accommodates important neural networks that regulate respiration, autonomic function, sleep, and arousal. Other physiological vulnerabilities in preterm infants relate to their autonomic nervous system and arousal mechanisms.